
Inhalt
- Ursachen
- Symptome
- Prüfungen und Tests
- Behandlung
- Selbsthilfegruppen
- Mögliche Komplikationen
- Wann wenden Sie sich an einen Arzt
- Verhütung
- Bilder
- Verweise
- Datum der Überprüfung 1/10/2018
Das Noonan-Syndrom ist eine Krankheit, die von Familien weitergegeben werden kann (vererbt). Dies führt dazu, dass sich viele Körperteile abnormal entwickeln.
Ursachen
Das Noonan-Syndrom ist mit Defekten in mehreren Genen verbunden. Im Allgemeinen werden bestimmte an Wachstum und Entwicklung beteiligte Proteine infolge dieser Genveränderungen überaktiv.
Das Noonan-Syndrom ist eine autosomal dominante Erkrankung. Dies bedeutet, dass nur ein Elternteil das nicht funktionierende Gen weitergeben muss, damit das Kind das Syndrom haben kann. Einige Fälle können jedoch nicht vererbt werden.
Symptome
Zu den Symptomen gehören:
- Verzögerte Pubertät
- Schräge oder weit aufgerissene Augen
- Hörverlust (variiert)
- Niedrige oder ungewöhnlich geformte Ohren
- Leichte geistige Behinderung (nur in etwa 25% der Fälle)
- Schlaffe Augenlider (Ptosis)
- Kleinwuchs
- Kleiner Penis
- Nicht angestiegene Hoden
- Ungewöhnliche Brustform (meistens eine versunkene Brust, genannt Pectus Baggatum)
- Gerippter und kurz erscheinender Hals
Prüfungen und Tests
Der Gesundheitsdienstleister führt eine körperliche Untersuchung durch. Dies kann Anzeichen für Herzprobleme aufweisen, die der Säugling von Geburt an hatte. Dazu können Lungenstenose und Vorhofseptumdefekt gehören.
Tests hängen von den Symptomen ab, können jedoch Folgendes umfassen:
- Thrombozytenzahl
- Blutgerinnungsfaktor-Test
- EKG, Röntgenaufnahme der Brust oder Echokardiogramm
- Hörtests
- Wachstumshormonspiegel
Gentests können helfen, dieses Syndrom zu diagnostizieren.
Behandlung
Es gibt keine spezifische Behandlung. Ihr Anbieter wird eine Behandlung vorschlagen, um die Symptome zu lindern oder zu behandeln. Wachstumshormon wurde bei einigen Menschen mit Noonan-Syndrom erfolgreich zur Behandlung von kurzer Körpergröße eingesetzt.
Selbsthilfegruppen
Die Noonan Syndrome Foundation ist ein Ort, an dem Menschen, die sich mit dieser Erkrankung befassen, Informationen und Ressourcen finden können.
Mögliche Komplikationen
Komplikationen können sein:
- Anormale Blutungen oder Blutergüsse
- Flüssigkeitsansammlungen im Körpergewebe (Lymphödem, zystisches Hygrom)
- Misserfolg bei Säuglingen
- Leukämie und andere Krebsarten
- Geringes Selbstwertgefühl
- Unfruchtbarkeit bei Männern, wenn beide Hoden unentschieden sind
- Probleme mit der Struktur des Herzens
- Kurze Höhe
- Soziale Probleme aufgrund körperlicher Symptome
Wann wenden Sie sich an einen Arzt
Dieser Zustand kann bei frühen Säuglingsuntersuchungen festgestellt werden. Für die Diagnose des Noonan-Syndroms wird häufig ein Genetiker benötigt.
Verhütung
Paare mit einer familiären Vorgeschichte des Noonan-Syndroms möchten möglicherweise eine genetische Beratung in Betracht ziehen, bevor sie Kinder bekommen.
Bilder
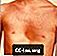
Pectus Exklatum
Verweise
Ali O, Donohoue, PA. Unterfunktion der Hoden. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, Hrsg. Nelson Lehrbuch der Pädiatrie. 20. ed. Philadelphia, PA: Elsevier; 2016: Kapitel 583.
Cooke DW, Divall SA, Radovick S. Normales und anormales Wachstum bei Kindern. In: Melmed S., KS Polonsky, PR Larsen, HM HM Kronenberg, Hrsg. Williams Lehrbuch der Endokrinologie. 13. Auflage Philadelphia, PA: Elsevier; 2016: Kapitel 24.
Madan-Khetarpal S, Arnold G. Genetische Störungen und dysmorphe Zustände. In: Zitelli BJ, McIntire SC, Nowalk AJ, Hrsg. Zitelli und Davis Atlas der pädiatrischen körperlichen Diagnose. 7. ed. Philadelphia, PA: Elsevier; 2018: Kap. 1
Datum der Überprüfung 1/10/2018
Aktualisiert von: Anna C. Edens Hurst, MD, MS, Assistenzprofessorin für medizinische Genetik, Universität Alabama in Birmingham, AL, Birmingham. Überprüfung durch das VeriMed Healthcare Network. Ebenfalls besprochen von David Zieve, MD, MHA, Ärztlicher Direktor, Brenda Conaway, Leitender Direktor und der A.D.A.M. Redaktion.