
Inhalt
- Ursachen
- Symptome
- Prüfungen und Tests
- Behandlung
- Selbsthilfegruppen
- Ausblick (Prognose)
- Wann wenden Sie sich an einen Arzt
- Verhütung
- Alternative Namen
- Bilder
- Verweise
- Überprüfungsdatum 15.10.2014
Die Niemann-Pick-Krankheit (NPD) ist eine Gruppe von Krankheiten, die durch Familien vererbt werden (vererbt), bei denen sich Fettstoffe, sogenannte Lipide, in den Zellen der Milz, der Leber und des Gehirns ansammeln.
Es gibt drei häufige Formen der Krankheit:
- Tippe A
- Typ B
- Geben Sie C ein
Jeder Typ beinhaltet verschiedene Organe. Es kann das Nervensystem und das Atmen betreffen oder nicht. Jeder kann verschiedene Symptome verursachen und kann zu verschiedenen Zeitpunkten im Leben auftreten.
Ursachen
Die NPD-Typen A und B treten auf, wenn die Zellen im Körper kein als Säure-Sphingomyelinase (ASM) bezeichnetes Enzym besitzen. Diese Substanz hilft, eine Fettsubstanz namens Sphingomyelin abzubauen (umzuwandeln), die in jeder Körperzelle vorkommt.
Wenn ASM fehlt oder nicht richtig funktioniert, baut sich Sphingomyelin in den Zellen auf. Das tötet Ihre Zellen und macht es den Organen schwer, richtig zu arbeiten.
Typ A kommt in allen Rassen und Ethnien vor. Es ist häufiger in der aschkenasischen (osteuropäischen) jüdischen Bevölkerung.
Typ C tritt auf, wenn der Körper Cholesterin und andere Fette (Lipide) nicht ordnungsgemäß abbauen kann. Dies führt zu zu viel Cholesterin in der Leber und Milz und zu viel anderen Lipiden im Gehirn. Typ C ist am häufigsten bei Puerto Ricanern spanischer Abstammung.
Typ C1 ist eine Variante von Typ C. Es handelt sich um einen Defekt, der die Cholesterinbewegung zwischen den Gehirnzellen beeinträchtigt. Dieser Typ wurde nur bei kanadischen Leuten in Yarmouth County, Nova Scotia, gesehen.
Symptome
Die Symptome variieren. Andere Gesundheitszustände können ähnliche Symptome verursachen. Die frühen Stadien der Krankheit können nur einige Symptome verursachen. Eine Person kann niemals alle Symptome haben.
Typ A beginnt normalerweise in den ersten Lebensmonaten. Symptome können sein:
- Schwellung des Bauches (Bauchbereich) innerhalb von 3 bis 6 Monaten
- Kirschroter Fleck im Augenhintergrund (auf der Netzhaut)
- Schwierigkeiten beim Füttern
- Verlust der frühen motorischen Fähigkeiten (wird mit der Zeit immer schlechter)
Typ-B-Symptome sind normalerweise milder. Sie treten in der späten Kindheit oder im Jugendalter auf. Bei kleinen Kindern kann es zu einer Abdominalschwellung kommen. Es gibt fast keine Einbindung des Gehirns und des Nervensystems, wie beispielsweise den Verlust motorischer Fähigkeiten. Bei einigen Kindern kann es zu wiederholten Infektionen der Atemwege kommen.
Typ C und C1 betreffen in der Regel Kinder im Schulalter. Es kann jedoch zu jeder Zeit zwischen der frühen Kindheit und dem Erwachsenenalter auftreten. Symptome können sein:
- Schwierigkeiten beim Bewegen der Gliedmaßen, die zu instabilem Gang, Unbeholfenheit und Gehproblemen führen können
- Vergrößerte Milz
- Vergrößerte Leber
- Gelbsucht bei (oder kurz nach) Geburt
- Lernschwierigkeiten und geistiger Verfall
- Anfälle
- Verschwommene, unregelmäßige Rede
- Plötzlicher Verlust des Muskeltonus, der zu Stürzen führen kann
- Zittern
- Mühe, die Augen auf und ab zu bewegen
Prüfungen und Tests
Ein Blut- oder Knochenmarkstest kann durchgeführt werden, um die Typen A und B zu diagnostizieren. Der Test kann erkennen, wer die Krankheit hat, aber es ist nicht ersichtlich, ob Sie Träger sind. DNA-Tests können durchgeführt werden, um Träger der Typen A und B zu diagnostizieren.
In der Regel wird eine Hautbiopsie durchgeführt, um die Typen C und D zu diagnostizieren. Der Arzt überwacht, wie die Hautzellen Cholesterin wachsen, sich bewegen und lagern. DNA-Tests können auch durchgeführt werden, um nach den 2 Genen zu suchen, die diese Art der Erkrankung verursachen.
Andere Tests könnten umfassen:
- Knochenmark Aspiration
- Leberbiopsie (in der Regel nicht erforderlich)
- Spaltlampen-Augenuntersuchung
- Tests zur Überprüfung des ASM-Levels
Behandlung
Derzeit gibt es keine wirksame Behandlung für Typ A.
Knochenmarktransplantationen können für Typ B versucht werden. Die Forscher untersuchen weiterhin mögliche Behandlungen, einschließlich Enzymersatz und Gentherapie.
Für die Symptome des Nervensystems des Typs C steht ein neues Medikament namens Miglustat zur Verfügung.
Hoher Cholesterinspiegel kann mit einer gesunden, cholesterinarmen Diät oder Medikamenten behandelt werden. Die Forschung hat jedoch nicht gezeigt, dass diese Methoden die Krankheit daran hindern, sich zu verschlechtern oder den Cholesterinabbau in den Zellen zu verändern. Arzneimittel stehen zur Verfügung, um viele Symptome zu kontrollieren oder zu lindern, wie zum Beispiel plötzlicher Verlust des Muskeltonus und Anfälle.
Selbsthilfegruppen
Diese Organisationen bieten Unterstützung und weitere Informationen zur Niemann-Pick-Krankheit:
- Nationales Institut für neurologische Erkrankungen und Schlaganfälle - www.ninds.nih.gov/Disorders/All-Disorders/Niemann-Pick-Disease-Information-Page
- National Niemann-Pick Disease Foundation - nnpdf.org
- Nationale Organisation für seltene Erkrankungen - rarediseases.org/rare-diseases/niemann-pick-disease-type-c
Ausblick (Prognose)
NPD Typ A ist eine schwere Erkrankung. Normalerweise führt dies im Alter von 2 oder 3 Jahren zum Tod.
Personen mit Typ B können bis in die späte Kindheit oder das Erwachsenenalter hineinleben.
Ein Kind, das vor dem 1. Lebensjahr Zeichen des Typs C aufweist, wird möglicherweise nicht bis zum Schulalter alt. Diejenigen, die nach dem Schuleintritt Symptome zeigen, können im mittleren bis späten Alter leben. Einige leben in ihren 20ern.
Wann wenden Sie sich an einen Arzt
Vereinbaren Sie einen Termin mit Ihrem Provider, wenn Sie eine Familienanamnese mit der Niemann-Pick-Krankheit haben und planen, Kinder zu bekommen. Eine genetische Beratung und ein Screening werden empfohlen.
Rufen Sie Ihren Anbieter an, wenn bei Ihrem Kind Symptome dieser Krankheit auftreten, einschließlich:
- Entwicklungsprobleme
- Fütterungsprobleme
- Schlechte Gewichtszunahme
Verhütung
Alle Arten von Niemann-Pick sind autosomal rezessiv. Dies bedeutet, dass beide Eltern Träger sind. Jedes Elternteil besitzt 1 Kopie des abnormen Gens, ohne Anzeichen der Krankheit selbst zu haben.
Wenn beide Eltern Träger sind, besteht eine 25% ige Chance, dass ihr Kind die Krankheit hat, und eine 50% ige Chance, dass ihr Kind Träger wird.
Carrier-Detection-Tests sind nur möglich, wenn der Gendefekt erkannt wird. Die bei den Typen A und B auftretenden Mängel wurden gut untersucht. DNA-Tests für diese Formen von Niemann-Pick sind verfügbar.
Bei vielen Menschen mit Typ C wurden genetische Defekte in der DNA identifiziert. Möglicherweise können auch Personen diagnostiziert werden, die das abnormale Gen tragen.
Einige Zentren bieten Tests an, um ein Baby im Mutterleib zu diagnostizieren.
Alternative Namen
NPD; Sphingomyelinase-Mangel; Lipid-Speicherstörung - Niemann-Pick-Krankheit; Lysosomale Speicherkrankheit - Niemann-Pick
Bilder
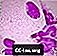
Niemann-Pick schaumige Zellen
Verweise
Kliegman RM, Stanton BF, St. Geme JW, Schor NF. Defekte im Stoffwechsel von Lipiden. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, Hrsg. Nelson Lehrbuch der Pädiatrie. 20. ed. Philadelphia, PA: Elsevier; 2016: Kapitel 86.
Patterson MC, Hendriksz CJ; NP-C-Richtlinienarbeitsgruppe, et al. Empfehlungen für die Diagnose und Behandlung der Niemann-Pick-Krankheit Typ C: ein Update. Mol Genet Metab. 2012; 106 (3): 330-344. PMID: 22572546 www.ncbi.nlm.nih.gov/pubmed/22572546.
Turnpenny PD, Ellard S. Angeborene Stoffwechselstörungen. In: Turnpenny PD, Ellard S, Hrsg. Emerys Elemente der medizinischen Genetik. 15. ed. Philadelphia, PA: Elsevier; 2017: Kapitel 18.
Überprüfungsdatum 15.10.2014
Aktualisiert von: Anna C. Edens Hurst, MD, MS, Assistenzprofessorin für medizinische Genetik, Universität Alabama in Birmingham, AL, Birmingham. Überprüfung durch das VeriMed Healthcare Network. Ebenfalls besprochen von David Zieve, MD, MHA, Ärztlicher Direktor, Brenda Conaway, Leitender Direktor und der A.D.A.M. Redaktion.